The code described above, being a relatively limited extension of the code in ref. [4], was used for large-scale production on the 128-node CM-5E at the JRCAT computer system in Tsukuba. Scientific results of using the code for studies of kinematic generation of dislocations in multi-grained copper crystals under strain have been reported in ref. [6].
Here we present performance results for this code on a typical dataset
from ref. [7], which can be compared to the results of our
previous paper[4]. A copper cluster with 178.219 atoms was
simulated using a spatial grid of 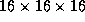 cells. A
near-melting-point temperature of about 1400 K was used as a rather
difficult test-case, giving rise to significant diffusion of atoms and
hence frequent updates of the neighbor-list and migration of atoms
between cells.
cells. A
near-melting-point temperature of about 1400 K was used as a rather
difficult test-case, giving rise to significant diffusion of atoms and
hence frequent updates of the neighbor-list and migration of atoms
between cells.
The performance of the new algorithm compared to the previous one[4] is shown in Table 1:
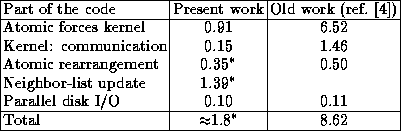
Table 1. Elapsed time (seconds) for a single MD time-step,
as measured on a 128-node CM-5E.
(* updates only performed every 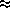 3 timesteps, see the text below)
3 timesteps, see the text below)
Since the algorithms with/without neighbor-lists differ significantly, one cannot compare all items in the table directly. However, the atomic forces kernel is now faster by a factor of more than 7 owing to the introduction of neighbor-lists. The kernel's computational speed is 5.1 GFLOPS, compared to the 11.7 GFLOPS for the ``old'' code[4], the difference being mainly due to ineffective code for indirect memory addressing through FORALL statements. Thus, the old code was more efficient, but due to the algorithm used, it still performed much less useful work per unit of time than the code implementing the present algorithm. The 5.1 GFLOPS corresponds to 25% of the CM-5E vector-units' theoretical peak performance, and this a very good absolute performance number.
The parallel load-balance due to fluctuations in atomic density is about the same for the new and old algorithms, and presents no problem for the present metallic solids. The system simulated in this work contained a significant fraction of cells containing vacuum (no atoms), whereas the present grid-based algorithms are more efficient for ``full'' systems such as bulk materials. There are imbalances in the present system's neighbor-lists: A neighbor-list contains up to about 83 atoms with an average of 68 atoms, when a skin-radius of 10% of the cutoff-radius is used. In a data-parallel code the longest neighbor-list determines the loop-length, so we have only about 80% load-balance here.
The kernel: communication item in Table 1 accounts for the data copying sketched in Fig. 1, constituting only 14% of the total force computation time. The parallel disk I/O using the CM-5E parallel SDA disks is a negligible part of the MD timestep.
The combined atomic rearrangement and neighbor-list updates require a time of about 1.6 times that of the the force computation. Fortunately, the neighbor-list is usually performed every 5-20 MD timesteps. In the present system at near-melting, however, updates are done about every 3 MD timesteps. This leads to an average MD timestep of about 1.8 seconds, or almost 5 times faster than the ``old'' algorithm for the same problem. For lower temperatures the neighbor-list updates would be less frequent, and the algorithmic speedup would be larger.